INFORMATIONS IMPORTANTES :
Dans le cadre de son nouveau plan stratégique, l’IPG a révisé l’ensemble de ses demandes d’analyse en y intégrant les toutes dernières évolutions médicales, une plus grande ergonomie ainsi qu’un respect strict de la législation en vigueur.
Vous les retrouverez en cliquant sur le bouton « DEMANDE D’ANALYSES ». Complémentairement, l’IPG assure une plus grande facilité d’accès à ses protocoles médicaux NIPT, HPV, Cyto et Anapath grâce à une publication, pour les médecins et les patients, de ses compte-rendus sur les Réseaux de Santé Wallon rsw.be et Bruxellois brusselshealthnetwork.be .
Hybridation Comparative de Genome sur micropuce (CGH microarray)
1. Principe de la méthode
La CGH sur micropuce, également appelée « caryotype moléculaire » permet d’explorer simultanément et sur tout le génome les déséquilibres chromosomiques (gain/perte) entre l’ADN d’une référence et celui d’un patient.
Le principe de la méthode est relativement simple: l’ADN du patient à tester est fragmenté et marqué par un fluorophore (Cyanine-5 dCTP donnant une couleur virtuelle rouge) et l’ADN d’un individu de référence est également fragmenté et marqué avec un autre fluorophore (Cyanine-3 dCTP de couleur virtuelle verte). Ces ADN marqués sont mélangés et co-hybridés en quantité égale sur une micropuce. Sur celle-ci, se trouve une série de spots d’ADN (plusieurs milliers). Chacun de ces spots est constitué d’un dépôt de fragments d’ADN spécifiques d’une région particulière du génome humain. Ces fragments correspondent à des oligo-nucléotides synthétisés in situ directement sur la micropuce.
Les ADN marqués vont s’hybrider avec la séquence d’ADN complémentaire présente sur la micropuce. Il doit donc s’établir un équilibre entre les molécules d’ADN marquées du patient et de la référence pour l’association avec la molécule d’ADN cible présente sur la micropuce. La mesure du rapport de fluorescence au niveau des différents spots de la micropuce permet de calculer un rapport du nombre de copies pour de multiples points dans le génome et déterminer ainsi les régions présentant un nombre de copies anormal (délétion ou duplication) par rapport à un individu de référence. Une transformation de ce rapport de fluorescence en logarithme en base 2 est effectuée. Ces valeurs sont placées sur un graphique en face de leur position chromosomique. Ainsi, une valeur de 0 indique une égalité des intensités des deux fluorochromes. Pour une délétion, les valeurs observées sont négatives et pour une duplication, ces valeurs sont positives.
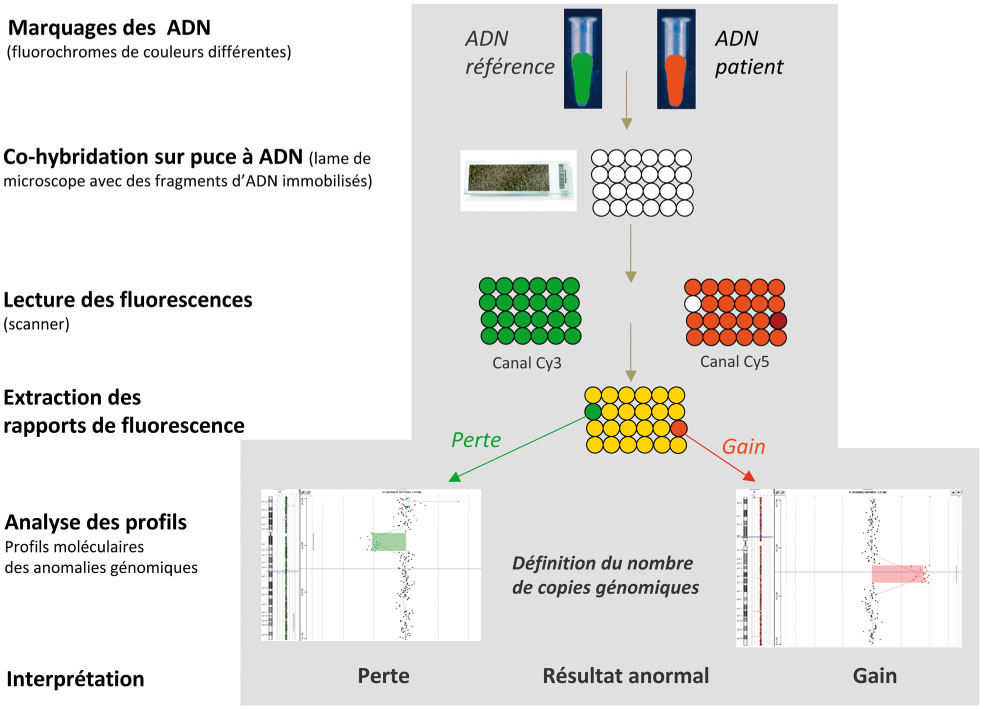
Image représentant les différentes étapes la technique CGH.
2. Avantages/Limitations:
L’avantage de la technique est sa capacité de localiser et d’identifier simultanément et sur l’ensemble du génome d’un individu des déséquilibres génomiques avec une résolution de 25 à 1000 fois supérieure au caryotype standard. La limite de résolution est fonction du nombre de sondes présentes sur la micropuce, de leur localisation et de la densité de celles-ci (régions ciblées à plus haute densité). En effet, seuls les réarrangements situés dans des régions couvertes par des sondes de la micropuce peuvent être identifiés.
Si la CGH est en mesure de mettre en évidence des réarrangements non équilibrés (délétion, duplication, amplification), elle ne peut cependant pas mettre en évidence les réarrangements équilibrés (translocations réciproques, inversions, insertions équilibrées), les disomies uniparentales, les polyploïdies et les mutations ponctuelles. Enfin les déséquilibres en mosaïques seront difficilement mis en évidence ; en particulier si le pourcentage de mosaïque est faible.
3. Les formats de micropuces utilisés à l’IPG
Différents formats de micropuces existent selon le nombre d’analyses pouvant être effectuées, le nombre de sondes oligonucléotidiques se trouvant sur celles-ci et la résolution désirée : au plus la micropuce contient de spots, au plus la résolution de celle-ci est élevée.
3.1. Secteur post-natal (CGH ISCA v2, 4x180K)
- L’ADN du patient est analysé sur une micropuce AGILENT couvrant tous les chromosomes (Numéro de catalogue AGILENT G4826A ; Numéro AMADID 31748 ; 146431 sondes uniques), avec une densité accrue dans des régions ou des gènes associés à des pathologies génétiques connues. Le choix des sondes a été effectué par un consortium international de laboratoire de cytogénétique (ISCA : International Standards for Cytogenomic Arrays Consortium). La CGH a été effectuée avec une polarité normale (l’ADN du patient a été marqué avec la Cyanine-5 et l’ADN de référence avec la Cyanine-3). L’ADN de référence utilisé est un ADN génomique commercial fourni par Promega®. Les régions aberrantes sont mises en évidence avec l’algorithme ADM2 dans le logiciel CytoGenomics® ou Genomic Workbench®, seuil de 6.0.
- La DLRSpread : “Derivative Log Ratio Spread » mentionnée dans chaque compte-rendu est une statistique donnant une estimation de la dispersion des valeurs de log2ratio en se basant sur les écarts entre les valeurs successives de log2ratio le long des chromosomes. Les valeurs attendues sont : Excellent : <0.2 ; Bon : 0.2-0.3 ; Faible : >0.3. Les positions sur le génome humain se basent sur la séquence du génome humain hg19 qui est la séquence publiée en février 2009 (« assembly » hg19, build GRCh37).
- Les données sont filtrées avant analyse et seuls les réarrangements impliquant 3 sondes consécutives ou plus sont pris en compte. Dès lors, la résolution théorique attendue est de l’ordre de 44 kb. Les aberrations localisées au niveau de polymorphismes connus du nombre de copies (Copy Number Variant : CNV) ; http://projects.tcag.ca/variation/ et notre base de donnée IPG en interne ne sont pas repris dans le compte-rendu.
- les réarrangements chromosomiques connus pour être associés avec le retard mental, un retard de développement ou des malformations congénitales peuvent être directement considérés comme responsables de la symptomatologie du patient. Par contre pour les réarrangements non rapportés dans la littérature, une analyse des parents par FISH ou par d’autres méthodes moléculaires est toujours recommandée pour aider à faire la distinction entre un polymorphisme rare hérité et un réarrangement pathologique qui puisse expliquer la symptomatologie du patient. Il est également important d’être conscient que les analyses par CGH sur micropuce peuvent mettre en évidence des réarrangements chromosomiques sans rapport avec les motifs de la consultation initiale mais révéler une susceptibilité à certaines pathologies, le portage d’un déterminant pathologique ou la présence d’un réarrangement dont la signification pathologique n’est pas claire aujourd’hui mais qui pourrait se révéler être pathologique ultérieurement. Il revient au clinicien d’en informer son patient et de déterminer s’il souhaite être informé de ces résultats inattendus.
3.2. Secteur prénatal (CGH ISCA v2, 8x60K)
- L’ADN est analysé sur une micropuce AGILENT couvrant tous les chromosomes (Numéro de catalogue AGILENT G4426B ; Numéro AMADID 24612 ; 59090 sondes uniques), avec une densité accrue dans des régions ou des gènes associés à des pathologies génétiques connues. Le choix des sondes a été effectué par un consortium international de laboratoire de cytogénétique (ISCA : International Standards for Cytogenomic Arrays Consortium). La CGH a été effectuée avec une polarité normale (l’ADN du patient a été marqué avec la Cyanine-5 et l’ADN de référence avec la Cyanine-3). L’ADN de référence utilisé est un ADN génomique commercial fourni par Promega®. Les régions aberrantes sont mises en évidence avec l’algorithme ADM1 dans le logiciel CytoGenomics® ou Genomic Workbench®, seuil de 6.0.
- La DLRSpread : “Derivative Log Ratio Spread » mentionnée dans chaque compte-rendu est une statistique donnant une estimation de la dispersion des valeurs de log2ratio en se basant sur les écarts entre les valeurs successives de log2ratio le long des chromosomes. Les valeurs attendues sont : Excellent : <0.2 ; Bon : 0.2-0.3 ; Faible : >0.3. Les positions sur le génome humain se basent sur la séquence du génome humain hg19 qui est la séquence publiée en février 2009 (« assembly » hg19, build GRCh37).
- Les données sont filtrées avant analyse et seuls les réarrangements impliquant 3 sondes consécutives ou plus sont pris en compte. Dès lors, la résolution théorique attendue est de l’ordre de 111 kb. Les aberrations localisées au niveau de polymorphismes connus du nombre de copies (Copy Number Variant : CNV) ; http://projects.tcag.ca/variation/ et notre base de donnée IPG en interne ne sont pas repris dans le compte-rendu.
- Les aberrations non interprétables en fonction de l’état des connaissances scientifiques au moment de l’analyse ne sont pas mentionnées. A cet égard, un consensus des 8 centres de Génétique Humaine Belge a été élaboré sous forme de guidelines, reprenant entre autres les CNV associés à des facteurs de susceptibilités. Le document est consultable à l’adresse suivante : http://www.beshg.be/download/guidelines/Guidelines_Prenatal_array_2013.pdf
3.3. Produits de fausses couches (Custom design CGH+SNP8x60k)
- L’ADN extrait de la fausse-couche est analysé sur une micropuce AGILENT couvrant tous les chromosomes (26697 sondes CGH uniques réparties sur l’ensemble des chromosomes). La CGH a été effectuée avec une polarité normale (l’ADN de la fausse-couche a été marqué avec la Cyanine-5 et l’ADN de référence avec la Cyanine-3). Etant donné que les triploïdies ne peuvent pas être mises en évidence par CGH, une détermination du génotype d’un ensemble de 26415 SNP (Single Nucléotide Polymorphism) est effectuée en parallèle. Cette analyse permet d’identifier les fausses couches polyploïdes. L’ADN de référence utilisé est un ADN de référence de génotype connu, de sexe masculin fourni par Agilent®. Les régions aberrantes sont mises en évidence avec l’algorithme ADM2 dans le logiciel CytoGenomics® ou Genomic Workbench®.
- La DLRSpread : “Derivative Log Ratio Spread » mentionnée dans chaque compte-rendu est une statistique donnant une estimation de la dispersion des valeurs de log2ratio en se basant sur les écarts entre les valeurs successives de log2ratio le long des chromosomes. Les valeurs attendues sont : Excellent : <0.2 ; Bon : 0.2-0.3 ; Faible : >0.3. Les positions sur le génome humain se basent sur la séquence du génome humain hg19 qui est la séquence publiée en février 2009 (« assembly » hg19, build GRCh37)
-Seules les aberrations de plus de 5 Mb ou de pathogénicité reconnue sont rapportées. Les aberrations localisées au niveau de polymorphismes du nombre de copies connus (Copy Number Variation : CNV ; http://projects.tcag.ca/variation/ et notre base de donnée interne à l’IPG) ne sont pas reprises dans les rapports. Les aberrations non interprétables en fonction de l’état des connaissances scientifiques au moment de l’analyse ne sont pas mentionnées.